پیشبینی محاسباتی واریانتهای ژنتیکی انسانی در ژنوم موش
موضوعات
- ژنتیک سرطان
- ژنومیک سرطان
- پایگاههای داده ژنتیکی
- غربالگری با توان بالا
- نرمافزار
چکیده
طراحی مدلهای موش مهندسیشده ژنتیکی میتواند از یک مسیر محاسباتی برای پیشبینی واریانتهای ژنتیکی موش که توالی و اثرات عملکردی واریانتهای بیماری انسانی را بازتاب میدهند، بهرهمند شود. در اینجا H2M (human-to-mouse) را معرفی میکنیم که این کار را با ترکیب تجزیه و تحلیلهای نقشهبرداری واریانتهای موش‑به‑انسان و پارالوگ‑به‑پارالوگ و ابزارهای ویرایش ژنوم انجام میدهد. ما یک پایگاه داده حاوی بیش از ۳ میلیون جفت جهش معادل انسانی‑موش و کتابخانههای ویرایش پایه و ویرایش اصلی برای مهندسی ۴٬۹۴۴ جفت واریانت ارائه میدهیم.
بخش اصلی
یکی از اهداف اصلی ژنتیک انسان، درک این است که چگونه واریانتهای ژنتیکی بر فنوتیپهای سلولی و مولکولی که پایهگذار بیماریهای انسانی هستند، اثر میگذارند. مدلهای موش مهندسیشده ژنتیکی (GEMMs) بهطور گستردهای برای مطالعه آسیبهای ژنتیکی مرتبط با بیماریهایی مانند سرطان به دلیل شباهت ژنتیکی وسیع با انسان و ارتباط فیزیولوژیکی مورد استفاده قرار میگیرند1,2,3,4. فناوریهای ویرایش ژنوم نیز بهطور فزایندهای در GEMMs بهکار گرفته میشوند تا درک بیماریهای انسانی تسریع شود5,6,7,8.
ناسازگاریهای ژنتیکی خاص گونه، توسعه و ارزیابی GEMMs را برای مطالعهٔ تنوع ژنتیکی انسانی و تفسیر اثرات زیستی پیچیده میسازد9. نقشهبرداری پیچیدهٔ غیرخطی ارگونومهای ژنی میتواند یافتن مکانهای ارگونوم که بتوان در موشها مهندسی کرد را دشوار سازد. اثرات تغییر توالیهای ارگونوم ممکن است بسته به زمینهٔ محلی توالی متفاوت باشد. واریانتهای موجود در سایتهای محافظتشده نیز ممکن است به دلیل تفاوتهای بینگونهای نقشهای متفاوتی در انسان و موش داشته باشند.
منابع ژنتیکی موجود و ابزارهای محاسباتی میتوانند کمک کنند، اما همچنان نیاز به پلتفرمهای یکپارچهای وجود دارد که فرهنگلغتهای جامع از واریانتهای ژنتیکی بین گونهها را برای مهندسی و مطالعهٔ تغییرات با توالی و/یا اثر عملکردی یکسان فراهم سازند. تجزیه و تحلیل نظاممند واریانتهای ارگونوم موش با ویرایش ژنوم با ابزارهای پیشبینی خودکار و کاربرپسند تسهیل میشود که نیازی به جستجوی دستی پرخطا در منابع مختلف ندارند. نتایج نیز باید استاندارد شوند تا امکان تجزیه و تحلیلهای بعدی مانند طراحی رناهای راهنما (guide RNA) و پیشبینی عملکرد پاتولوژیک فراهم گردد.
ما H2M (human-to-mouse؛ https://github.com/kexindon/h2m-public) را توسعه دادیم، یک مسیر محاسباتی که دادههای تنوع ژنتیکی انسانی را پردازش میکند تا پیامدهای واریانتهای معادل موش را مدلسازی و پیشبینی کند و به تدوین استراتژیهای مهندسی دقیق برای وارد کردن جهشهای متناظر در موشها کمک نماید. H2M با استفاده از دادههای واریانتهای ژنتیکی بهعنوان ورودی، بهصورت نظاممند واریانتهای ارگونوم را در میان هزاران جهش شناسایی، مدلسازی و تصویری میسازد. هرچند ما در اینجا کاربرد آن را برای تجزیه و تحلیلهای human‑to‑mouse و mouse‑to‑human نشان میدهیم، H2M برای هر گونهای که ژنوم مرجع توالیدار داشته باشد، سازگار است.
H2M چهار گام اصلی را اجرا میکند: (1) پرسوجو از ژنهای ارگونوم؛ (2) تراز کردن رناهای وحشی یا پپتیدها؛ (3) شبیهسازی جهشها؛ و (4) بررسی و مدلسازی اثرات عملکردی (شکل 1a و شکل دادههای تکمیلی 1). این ابزار از یک فهرست داخلی از همولوگهای موش و انسان10,11,12,13 (جدول تکمیلی 1) استفاده میکند تا جفتهای ژنی مورد علاقه را شناسایی کرده و سپس توالیهای کامل و تمام نسخههای رنا برای هر ژن را بازیابی نماید.
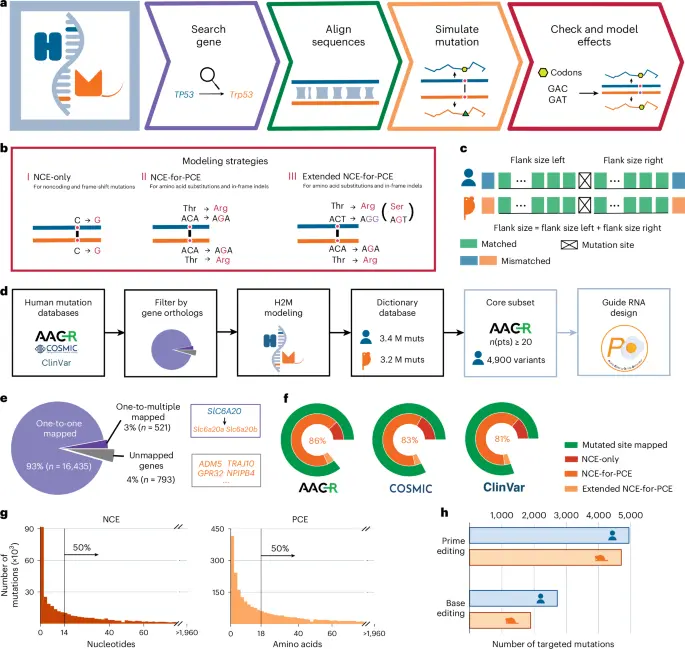
a, H2M چهار گام اصلی را اجرا میکند: (1) پرسوجو از ژنهای ارگونوم؛ (2) تراز کردن رناهای وحشی یا پپتیدها؛ (3) شبیهسازی جهشها؛ و (4) بررسی و مدلسازی اثرات عملکردی. b, H2M بر اساس اثر تغییر توالی ورودی، از سه استراتژی مدلسازی استفاده میکند. برای جهشهای غیرکدینگ و تغییر چارچوب، H2M از (I) مدلسازی NCE‑only برای شبیهسازی همان تغییر در سطح DNA بهره میبرد. برای جایگزینیهای آمینو اسید و ایندلها، H2M یا (II) NCE‑for‑PCE را به کار میگیرد اگر جهش DNA باعث تغییر آمینو اسید یکسان در هر دو ژنوم شود، یا (III) مدلسازی NCE‑for‑PCE گسترشیافته را اگر برای شبیهسازی تغییر آمینو اسید هدف به جهش DNA متفاوتی نیاز باشد. c, طرح کلی توالی اطراف (flank) برای محل جهش. اندازه flank بهعنوان مجموع طول نوکلئوتیدهای توافقی (برای واریانتهای غیرکدینگ) یا پپتیدها (برای واریانتهای کدینگ) در دو طرف مکان جهش تعریف میشود. d, نمودار گرافیکی تولید پایگاه داده H2M. M، میلیون؛ muts، جهشها. e, نمودار دایرهای نمایشدهندهٔ حضور ارگونومهای ژن موش برای ژنهای انسانی موجود در مجموعه دادهٔ ورودی. f, درصدهای جهشهای انسانی موجود در پایگاه داده H2M که میتوانند در ژنوم موش مدلسازی شوند، بر اساس منبع داده طبقهبندی شدهاند. g, توزیع اندازههای flank برای تمام واریانتهای انسانی در پایگاه داده H2M، جداگانه برای NCE (سمت چپ) برای جهشهای غیرکدینگ و PCE (سمت راست) برای جهشهای کدینگ. h, تعداد جهشهایی که در زیرمجموعه انتخابی پایگاه داده H2M قابلیت ویرایش اولیه (prime‑editing) و ویرایش پایه (base‑editing) دارند. NCE، اثر تغییر نوکلئوتیدی؛ PCE، اثر تغییر پپتیدی.
برای هر رنا، H2M اگزونها و اینترونها را شناسایی میکند، افراز RNA را شبیهسازی مینماید و توالیهای کامل رنا را بهدست میآورد. سپس اثرات عملکردی جهشهای هدف ژن را در سطح نوکلئوتید و پپتید شبیهسازی، بررسی و مدلسازی میکند. برای تعیین اینکه آیا جهشها به مناطق محافظتشدهٔ محلی نقشه میشوند یا نه، H2M با استفاده از الگوریتم نیدلمن‑ونش رناهای وحشی (برای جهشهای غیرکدینگ) یا توالیهای پپتید (برای جهشهای کدینگ) را تراز مینماید. اگر جهش انسانی یک مکان معادل در ژنوم موش داشته باشد، H2M از سه استراتژی مدلسازی استفاده میکند (شکل 1b و شکل دادههای تکمیلی 1).
برای تمامی ورودیها، H2M همان تغییر نوکلئوتیدی را در رناهای موش محاسبه میکند و NCE معادل (اثر تغییر نوکلئوتیدی) را خروجی میدهد، که بهعنوان تغییر در سطح DNA ناشی از یک جهش تعریف میشود (استراتژی I: مدلسازی NCE‑only؛ شکل دادههای تکمیلی 2a–c). چون همان تغییر نوکلئوتیدی در مکانهای معادل انسانی و موش همیشه به همان آمینو اسید منجر نمیشود، H2M اثرات تغییر توالی در سطح پروتئین (اثر تغییر پپتیدی، PCE) را برای واریانتهای کدینگ (یعنی تغییر آمینو اسید) نیز محاسبه میکند. برای در نظر گرفتن این تفاوتهای بالقوه، H2M تغییرات در سطح DNA ایجاد میکند که باید همان اثر کدینگ پروتئینی را در هر دو گونه تولید کنند. پس از شبیهسازی همان NCE در هر دو ژن و مقایسه تغییرات آمینو اسید بهدست آمده، H2M واریانت معادلی که هم NCE و هم PCE را بازتاب میدهد، نگه میدارد (استراتژی II: مدلسازی NCE‑for‑PCE؛ شکل دادههای تکمیلی 2d). در غیر این صورت، H2M سعی میکند معادلهای PCE گسترشیافته با NCEهای متفاوت بر پایهٔ افزونگی کدونها ارائه دهد (استراتژی III: مدلسازی NCE‑for‑PCE گسترشیافته؛ شکل دادههای تکمیلی 2e).
خروجی H2M شامل اطلاعات استاندارد فراوانی است که میتوان برای انواع مختلف تجزیه و تحلیلهای بعدی استفاده کرد (جدول دادههای تکمیلی 2). علاوه بر مختصات جهشها و تغییرات توالی در سطح DNA به فرمت MAF، H2M اثرات تغییر توالی در سطح رنا و پروتئین را با استفاده از نامگذاری استاندارد HGVS ارائه میدهد14.
مطالعات توالیگذاری ژنوم میلیونها جهش ژنتیکی انسانی در زمینههای سلولهای جنینی و سوماتیک را فهرست کردهاند. یک فهرست معادل موشانه میتواند برای پیشبینی اثرات این جهشها، تدوین استراتژیهای ساخت مدلهای جدید GEMM و تفسیر دادههای تجربی از مدلهای موجود ارزشمند باشد. با این هدف، ما پایگاه دادههای AACR‑GENIE15 و COSMIC را بررسی کردیم.